







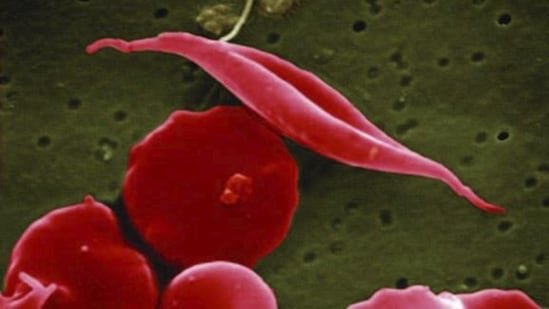
Sickle cell disease (SCD) is a blood disorder that causes red blood cells to become sickle-shaped, leading to chronic anemia, severe pain episodes, and life-threatening complications. In India, SCD remains a significant public health challenge, particularly among tribal population and few regional belts, emphasising the need for early diagnosis, comprehensive care, and greater awareness.
Accurate diagnosis of SCD is essential because early identification allows timely treatment, genetic counseling, and prevention of serious complications. The diagnosis is based on a combination of screening tests, hematological evaluation, haemoglobin analysis, and molecular methods. Modern guidelines emphasise that a definitive diagnosis should rely on identification of the abnormal haemoglobin pattern and, when required, confirmation by genetic testing.
The initial laboratory evaluation often begins with a complete blood count (CBC). Many patients with SCD show anemia, usually of a normocytic or mildly macrocytic type, along with an increased reticulocyte count due to ongoing hemolysis. However, CBC findings alone are not diagnostic because similar abnormalities may occur in other hemolytic disorders.
Examination of the peripheral blood smear provides valuable clues. Characteristic sickle-shaped red blood cells, target cells, polychromasia, and occasionally nucleated red blood cells may be observed. While these findings strongly suggest SCD, they are insufficient for confirmation because the number of sickled cells may vary and some patients, especially infants, may not show classic morphology.
Simple screening tests such as the sickling test and the haemoglobin solubility test detect the presence of haemoglobin S (HbS). These tests are inexpensive and useful in resource-limited settings. The solubility test is based on the reduced solubility of deoxygenated HbS, producing a turbid solution when HbS is present. However, these methods cannot distinguish between sickle cell trait and sickle cell disease. They may also yield unreliable results in newborns because of high foetal haemoglobin levels and therefore should never be used as the sole basis for diagnosis.
These are the two widely used methods for Hb electrophoresis.
High-performance liquid chromatography (HPLC) has become a preferred method in many centers because it not only identifies but also accurately quantifies different haemoglobin fractions. The technique separates hemoglobins based on their retention times and provides precise measurement of HbS, HbF, HbA₂, HbC, and other variants.
HPLC is particularly useful for newborn screening programs and for monitoring patients receiving therapies such as hydroxyurea or regular blood transfusions. Its high sensitivity and reproducibility make it one of the most powerful laboratory tools for haemoglobinopathy diagnosis. Nevertheless, certain variants may share similar retention times, and therefore HPLC findings occasionally require further confirmation by capillary electrophoresis or genetic testing.
Capillary electrophoresis is another highly accurate method that separates hemoglobin fractions using electrophoretic mobility in a capillary system. This test distinguish cases of co-inheritence of beta-thallasemia variant. Like HPLC, it is also a part of new born screening in many places.
New born screening: This is a simple test works on the principle of haemoglobin electrophoresis and needs only a few drops of blood from newborn’s heel. This can detect presence of haemoglobin S at very early stage and make early interventions possible, preventing complications and better prognosis.
Genetic analysis: These tests analyse the DNA and assist in identification of such abnormal gene which leads to formation of abnormal haemoglobin. Hence, genetic testing is very promising in confirming diagnosis in borderline and complex case like sicke-beta thalassemia. It helps in identifying the type of mutation and presence of co-inheritance of any other abnormal haemoglobin chain (alpha or beta chain variant), which might not be differentiated in hb electrophoresis. Genetic testing is also a part of pre-natal testing of foetus.
Pre-natal Testing: If both parents are known carriers, the unborn baby can be tested during pregnancy. The methods include Chorionic Villus Sampling (CVS) at 10–12 weeks, or amniocentesis at 15–18 weeks. These tests analyse the baby’s DNA and if the abnormal gene is inherited. This allows families to make informed decisions with full medical counselling and support.
Today, the most reliable diagnostic approach combines hematological assessment with haemoglobin characterisation by electrophoresis, HPLC, or capillary electrophoresis, followed by molecular confirmation whenever necessary. This integrated strategy ensures accurate diagnosis and appropriate clinical management of patients with sickle cell disease.
(The views expressed are personal)
This article is authored by Dr Namrata Singh, consultant pathologist, Dept of Haematology and HLA, Metropolis Healthcare Limited.








